Afecten menys d’
1
de cada 2.000 persones
Es coneixen més de
250
gens que hi tenen relació
Què són les distròfies de la retina?
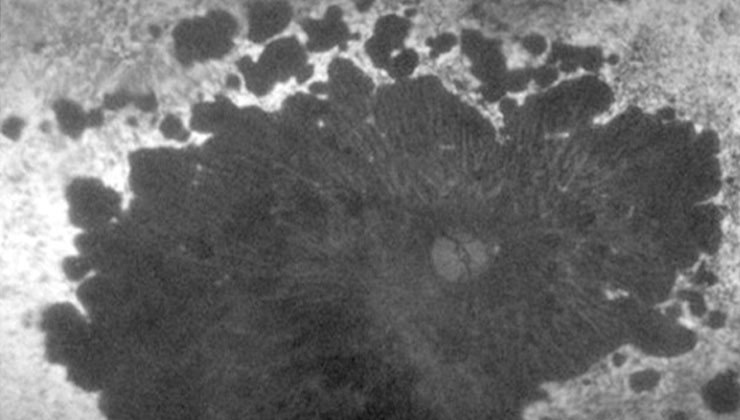
Les distròfies retinals són un conjunt heterogeni de malalties hereditàries que provoquen una pèrdua progressiva i severa de visió, atès que alteren l’anatomia i/o la funció de la retina.
Actualment no tenen cura, tot i que s’està investigant per poder tractar-les amb teràpies gèniques i cel·lulars en els propers anys.
Aquestes patologies poden causar un dany en les cèl·lules fotoreceptores, ja siguin, predominantment, els cons (responsables de la visió en detall i de color), els bastons (especialitzats en la visió nocturna i perifèrica) o tots dos alhora. És el cas de la malaltia de Stargardt, la retinosi pigmentària o la distròfia de cons-bastons, respectivament.
També hi ha certes distròfies hereditàries, com la retinòsquisi juvenil, la vitreoretinopatia exsudativa familiar o la síndrome de Stickler, en què es produeixen alteracions del vitri i de la retina. En altres, com la coroiderèmia, el problema de base és a la coroide, capa situada per sota de la retina.
La majoria de distròfies de retina són malalties localitzades exclusivament a l’ull, malgrat que de vegades poden associar-se a manifestacions extraoculars (síndrome d’Usher, síndrome de Bardet-Biedl), en el cas de les quals parlem de distròfies retinals sindròmiques.
Per la seva baixa prevalença, les distròfies de la retina són considerades malalties minoritàries (afecten menys d’1 de cada 2.000 persones).
Per què es produeixen?
Les distròfies de la retina tenen origen genètic, de manera que poden transmetre’s de generació en generació mitjançant diferents tipus d’herència.
- Herència dominant: acostuma haver-hi afectats en totes les generacions de la família, ja que les persones portadores de la mutació responsable de la malaltia la manifesten. Aquesta patologia es transmet aproximadament al 50% dels descendents, com la malaltia de Best.
- Herència recessiva: habitualment només hi ha familiars afectes en una generació, ja que els pacients portadors d’una mutació estan sans, mentre que els portadors de dues mutacions en el mateix gen desenvolupen la malaltia. Així ocorre en la malaltia de Stargardt.
- Herència lligada al cromosoma X: únicament els homes de la família pateixen la patologia, malgrat que les dones poden ser portadores de la mutació i transmetre la malaltia als fills nois amb un 50% de probabilitat. En aquest grup s’engloba, entre d’altres, la coroiderèmia.
La retinosi pigmentària, la distròfia de la retina més freqüent, és un exemple de patologia que es pot transmetre a través dels tres patrons d’herència mencionats, en funció del gen implicat.
La genètica de les distròfies de retina és complexa: una mateixa patologia pot estar causada per diversos gens i, a la vegada, un mateix gen pot estar relacionat amb diferents malalties. Actualment, s’han descrit més de 250 gens associats a les distròfies retinals, tot i que s’estima que encara en falten molts per identificar.
Com es poden prevenir?
La informació genètica continguda en l’ADN de cada persona és la que determina que es desenvolupi una distròfia de la retina i, per tant, aquestes patologies hereditàries no es poden prevenir. No obstant això, un aspecte en el qual sí que es pot incidir és la detecció precoç per part d’oftalmòlegs especialistes, mitjançant una revisió ocular completa que inclogui una bona anamnesi i una exploració exhaustiva del fons de l’ull, a més de tècniques complementàries, com l’OCT, l’autofluorescència o les proves electrofisiològiques.
Sovint, les distròfies retinals no són evidents fins a fases avançades, quan els símptomes es fan patents. Per poder anticipar-se a la malaltia i per predir-ne l’evolució, és de gran utilitat conèixer la causa molecular que la desencadena en cada pacient mitjançant el diagnòstic genètic, que també permet confirmar el diagnòstic clínic, identificar el patró d’herència i oferir consell genètic a la família. D’aquesta manera és possible indicar la probabilitat de transmetre la patologia i alertar els familiars portadors.
Així mateix, el diagnòstic genètic obre la porta a prevenir, aturar o revertir en el futur la pèrdua de visió dels pacients afectats, mitjançant el desenvolupament de noves teràpies individualitzades que estan en investigació.
Símptomes
Els símptomes, així com la rapidesa de l’evolució, varien segons el tipus de distròfia de retina i en funció de cada pacient. Tanmateix, els més comuns són:
- Pèrdua d’agudesa visual: dificultat per apreciar detalls i dur a terme tasques de precisió.
- Reducció del camp de visió: visió “en túnel” o aparició de “punts cecs” (escotomes).
- Ceguesa nocturna: problemes per veure-hi de nit o en ambients foscos i mala adaptació a situacions de poca lluminositat.
- Fotofòbia i enlluernaments: especials molèsties davant la llum i visió de “flaixos” o reflexos en condicions d’alta lluminositat. El pas d’un ambient clar a un entorn fosc –i viceversa– també genera problemes.
Altres senyals possibles poden ser la percepció deformada d’objectes (metamorfòpsia) o l’alteració en la percepció dels colors (discromatòpsia).
Tractaments associats
Les distròfies de la retina actualment no tenen cura, atesa la dificultat de regenerar les cèl·lules retinals afectades. El futur tractament d’aquestes patologies passa pel disseny i l’aplicació de noves teràpies gèniques i cel·lulars que permetin retornar visió o frenar-ne la pèrdua.
Actualment, s’estan fent passos importants en aquesta línia de recerca (des del 2018, s’ha començat a comercialitzar la primera terapia gènica als Estats Units, dissenyada per a un gen responsable d’ amaurosi congènita de Leber) i el Departament de Genètica de l’Institut també s’orienta a aquest repte amb diversos projectes promoguts per la Fundació IMO.
Per preparar els candidats a aquestes teràpies gèniques és fonamental disposar d’un diagnòstic genètic, que l’IMO ofereix de forma pionera.
En paral·lel, l’Institut també aposta per una altra via alternativa i complementària de tractament: la visió artificial, al desenvolupament de la qual hi contribueix participant en estudis amb nous models de xip de retina. És el cas de l’IRIS®II, que ha implantat en la primera pacient d’Europa.
Especialistes que tracten aquesta patologia











Preguntes freqüents dels nostres pacients
La retinitis pigmentària és la malaltia hereditària més freqüent de la retina. Es caracteritza per una degeneració progressiva de la retina. Actualment no té tractament. Per això, i per la seva gravetat, és una de les patologies oculars d’origen genètic sobre les quals s’està investigant més. La teràpia gènica està donant resultats molt esperançadors. Tot i que encara no hi ha tractament, els pacients amb retinosi podrien acudir al Departament de Genètica de l’IMO per fer els estudis que estan en procés.
Segons el que es desprèn de les conclusions del Congrés Internacional de Retina celebrat a l’IMO, estudis en fase I en humans sembla que ja demostren que l’ús de cèl•lules mare per reemplaçar cèl•lules danyades de la retina aconsegueix millorar l’agudesa visual dels pacients. Aquesta teràpia s’aplica en pacients que perden cèl•lules fotoreceptores i/o de l’epiteli pigmentari, un tipus de cèl•lules que no es regeneren i que són fonamentals per a la visió.
El que s’està aconseguint amb les noves teràpies és reemplaçar-les per cèl•lules mare embrionàries o pluripotencials extretes de la pell o d’altres parts de l’ull que, alterades, són capaces de desenvolupar la mateixa funció que les cèl•lules retinals danyades. En aquests moments, aquest tractament s’està aplicant en fase de proves i amb molt bons resultats a pacients amb distròfies retinals, retinosi pigmentària i DMAE.
Hi ha moltes malalties oculars que estan lligades a l’herència cromosòmica i fins i tot està determinat el tipus de defecte genètic. Malalties de qualsevol part de l’ull poden tenir caràcter hereditari. La més coneguda és la retinosi pigmentària.
Antelació
A vegades, els pacients volen fer-se tractaments per veure’s millor abans d’un esdeveniment o d’una ocasió especial. En aquests casos, en línies generals, se sol recomanar dur-los a terme amb una antelació mínima de tressetmanes, perquè:
- es puguin apreciar els resultats definitius,
- no hi hagi marques i
- tenir marge per fer retocs, en cas que siguin necessaris.
Tractaments quirúrgics
D’altra banda, els tractaments quirúrgics se solen fer amb una antelació mínima de 2 mesos per poder apreciar els resultats definitius. Generalmentsón cirurgies mínimament invasives, sense marques visibles, que tenen per objectiu mantenir l’harmonia del rostre del mateix pacienti allunyar-se de resultats massa artificials.
La millor època de l’any
La majoria de tractaments tant de medicina estètica com quirúrgics poden efectuar-se en qualsevol moment de l’any.
L’única excepció són els làsers (IPL i CO₂), els quals no s’apliquen en període estival, atèsque els raigs del sol incideixen de manera negativa en el postractament.
Descobreix aquí tots els tractamentsdel Departament d’Estètica.
Si no té gas ni oli de silicona pot dormir en qualsevol posició. Com que no hi ha cap element taponador (gas o oli de silicona), no és necessari que el pacient vigili la posició.
Potser t'interessa
IMO Institut de Microcirurgia Ocular
Josep María Lladó, 3
08035 Barcelona
Tel: 934 000 700
E-mail: informacion@imo.es
Veure mapa a Google Maps
En cotxe
Coordenades navegador GPS:
41º 24’ 38” N – 02º 07’ 29” E
Sortida 7 de la Ronda de Dalt (costat muntanya). La clínica compta amb un aparcament de més de 200 places.
En bus
Autobús H2: Rotonda de Bellesguard, parada 1540
Autobús 196: Josep Maria Lladó-Bellesguard, parada 3191
Autobusos H2, 123, 196: Ronda de Dalt – Bellesguard, parada 0071
Com arribar a IMO des de:
IMO Grup Miranza Madrid
C/ Valle de Pinares Llanos, 3
28035 Madrid
Tel: 910 783 783
Veure mapa en Google Maps
Transport públic
Metre Lacoma (línia 7)
Autobusos:
- Línies 49 i 64, parada “Senda de l’Infant”
- Línia N21, parada “Metre Lacoma”
Horaris
Atenció al pacient: de dilluns a divendres, de 8 h a 21 h
IMO Grup Miranza Andorra
Av. de les Nacions Unides, 17
AD700 Escaldes-Engordany, Andorra
Tel: (+376) 688 55 44
Veure mapa a Google Maps
IMO Manresa
C/ Carrasco i Formiguera, 33 (Baixos)
08242 – Manresa
Tel: 938 749 160
Veure el mapa a Google Maps
Transport públic
FGC. Línia R5 i R50 direcció Manresa. Estació: Baixador de Manresa
Horaris
De dilluns a divendres de 9:00 a 19:00h