
Fins un
50%
de probabilitat de transmetre la patologia per herència dominant
En marxa
2
projectes d’investigació sobre nous gens implicats
Més de
100
gens implicats i més de 2.000 mutacions identificades
Què és la retinosi pigmentària?
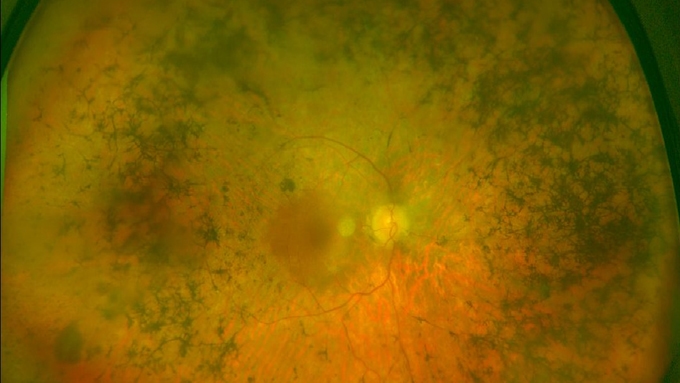
És una patologia hereditària que es caracteritza per la degeneració precoç i progressiva de les cèl·lules retinals, fonamentalment, les fotoreceptores: els bastons (visió nocturna) i els cons (percepció de colors i de detalls), que converteixen la llum en senyals que es transmeten al cervell.
La retinosi pigmentària s’engloba dins del grup de les malalties considerades minoritàries – afecta aproximadament 1 de cada 3.000 persones–, tot i que es tracta de la distròfia hereditària de la retina més freqüent.
Per què es produeix?
La retinosi pigmentària és una malaltia genètica de la retina.
Actualment, hi ha descrits més de 100 gens implicats i s’han identificat més de 2.000 mutacions. No obstant això, només expliquen una part dels casos de retinosi pigmentària i, en prop d’un 25-30% de les famílies afectades a les quals es fa un diagnòstic genètic, no s’aconsegueix identificar la causa molecular. Això vol dir que encara queden nous gens i mutacions per associar a la malaltia, un repte al qual s’orienta la investigació i per al qual la Fundació IMO té actualment 2 projectes en marxa al laboratori.
Herència de la retinosi pigmentària
La genètica és complexa en aquesta distròfia de la retina, ja que es pot transmetre a través de diferents patrons d’herència:
- Dominant: afectes en totes les generacions de la família.
- Recessiva: salta generacions. Hi ha afectes en una generació i, en les altres, membres portadors.
- Lligada al cromosoma X: afectes només homes i dones portadores.
Com es pot prevenir?

Atès que és d’origen genètic, no es pot evitar l’aparició de la retinosi pigmentària. No obstant això, detectar-la aviat permet anticipar-se a la malaltia i realitzar un bon seguiment oftalmològic dels pacients per controlar possibles problemes associats (per exemple, altres alteracions a la retina o cataracta).
Com a reforç del diagnòstic clínic, disposar d’un diagnòstic genètic que determini la causa molecular de la patologia és d’utilitat per a identificar-ne el patró d’herència i indicar la probabilitat de transmetre-la. A més, permet alertar familiars portadors i membres afectes que encara no l’han desenvolupat, però que poden manifestar-la en el futur.
En famílies amb antecedents de la malaltia, hi ha la possibilitat de fer un diagnòstic genètic preimplantacional per, mitjançant fecundació in vitro, transferir a l’úter matern embrions lliures de la mutació. D’altra banda, el diagnòstic genètic prenatal, a partir d’una mostra de líquid amniòtic obtinguda durant l’embaràs, permet detectar abans del naixement la presència de la mutació responsable de la retinosi pigmentària.
Símptomes
Tot i que la mutació genètica està present des del naixement, la retinosi pigmentària sol començar a donar els primers símptomes en l’adolescència. La pèrdua gradual de visió que provoca afecta generalment ambdós ulls, tot i que de forma desigual, i acostuma a ser lenta. Ara bé, la velocitat de progressió de la patologia i la severitat és molt variable, en funció de la mutació i del gen afectat en cada cas.
Aquests són alguns dels principals símptomes que experimenten els pacients:
- Ceguesa nocturna: la dificultat de visió en entorns poc il·luminats i la mala adaptació de la claredat a la foscor són sovint les primeres manifestacions de la retinosi pigmentària.
- Visió en túnel: la reducció del camp visual fa que els pacients vagin perdent visió perifèrica, per la qual cosa és habitual que pateixin cops i caigudes freqüents o que tinguin problemes per localitzar i agafar objectes al seu voltant. Per contra, la visió central es manté fins a fases tardanes de la malaltia.
- Enlluernaments: la percepció de “llampecs” i de “flaixos”, sobretot en condicions de molta lluminositat, resulta molt molesta i fa necessari l’ús d’ulleres de sol amb filtres especials.
- Disminució de l’agudesa visual: la capacitat de veure-hi amb nitidesa i de distingir detalls minva quan la patologia està evolucionada.
- Alteració en la percepció dels colors: també ocorre en casos avançats de retinosi pigmentària.
Tractaments associats
Actualment, no és possible aturar la pèrdua de visió que provoca la retinosi pigmentària (malgrat que sí que es pot aprofitar la resta visual dels pacients gràcies a ajudes de baixa visió).
Per donar amb un tractament efectiu, és una de les patologies oculars d’origen hereditari sobre les quals s’està fent més recerca, amb diverses línies d’estudi que esperen obtenir fruits en els propers anys:
Teràpies gèniques: estan en fase avançada d’investigació i consisteixen en la substitució del gen defectuós per un de sa per evitar que progressi la malaltia. Requereixen conèixer la causa molecular en cada pacient, de manera que només són aptes per a qui prèviament s’hagi identificat la mutació en el gen responsable mitjançant el diagnòstic genètic, imprescindible per poder dissenyar una estratègia terapèutica individualitzada.
Teràpies cel·lulars: basades en la introducció de cèl·lules sanes en el teixit retinal afectat, al qual s’han d’integrar (per complir amb les complexes funcions de les cèl·lules de la retina després d’haver-se cultivat al laboratori a partir de les cèl·lules mare obtingudes d’un altre teixit). Aquesta estratègia és independent de la causa molecular i, un cop aconsegueixin superar-les dificultats del procés, es podrà aplicar en estadis més severs de la patologia que les teràpies gèniques.
Xip de retina: la visió biònica ja fa possible la percepció d’estímuls lluminosos per facilitar la localització i la identificació d’objectes en persones invidents. S’està treballant en models que aportin cada vegada més resolució de les imatges i que permetin donar el salt definitiu d’aquesta tecnologia que estimula artificialment la retina.
Els especialistes de l’IMO estan involucrats tant en projectes de recerca que es duen a terme al laboratori de biologia molecular per establir les bases de l’aplicació de teràpies gèniques i cel·lulars, com en el desenvolupament de la visió artificial. En aquest sentit, l’Institut ha implantat el xip de retina IRIS®II, per primera vegada fora d’estudis a Europa, en una pacient amb retinosi pigmentària, a més de seguir involucrat en nous models i avenços.
Especialistes que tracten aquesta patologia











Preguntes freqüents dels nostres pacients
La retinitis pigmentària és la malaltia hereditària més freqüent de la retina. Es caracteritza per una degeneració progressiva de la retina. Actualment no té tractament. Per això, i per la seva gravetat, és una de les patologies oculars d’origen genètic sobre les quals s’està investigant més. La teràpia gènica està donant resultats molt esperançadors. Tot i que encara no hi ha tractament, els pacients amb retinosi podrien acudir al Departament de Genètica de l’IMO per fer els estudis que estan en procés.
Segons el que es desprèn de les conclusions del Congrés Internacional de Retina celebrat a l’IMO, estudis en fase I en humans sembla que ja demostren que l’ús de cèl•lules mare per reemplaçar cèl•lules danyades de la retina aconsegueix millorar l’agudesa visual dels pacients. Aquesta teràpia s’aplica en pacients que perden cèl•lules fotoreceptores i/o de l’epiteli pigmentari, un tipus de cèl•lules que no es regeneren i que són fonamentals per a la visió.
El que s’està aconseguint amb les noves teràpies és reemplaçar-les per cèl•lules mare embrionàries o pluripotencials extretes de la pell o d’altres parts de l’ull que, alterades, són capaces de desenvolupar la mateixa funció que les cèl•lules retinals danyades. En aquests moments, aquest tractament s’està aplicant en fase de proves i amb molt bons resultats a pacients amb distròfies retinals, retinosi pigmentària i DMAE.
És una tècnica diagnòstica per determinar les estructures patològiques i anormals que té la retina, tant als vasos com a les diferents capes que té. És vàlida per a la degeneració de la màcula, per a la retinopatia diabètica i per a moltes altres alteracions maculars i per a vasculopaties.
L’angiografía és una tècnica que serveix per delinear els casos de la retina o la coroide. Es fan servir diferents contrastos, normalment la fluoresceïna sòdica o el verd indocianina. Aquesta exploració també és molt útil per al diagnòstic d’altres afeccions de la retina, especialment de l’anomenat epiteli pigmentat. En general, les angiografies serveixen per estudiar múltiples malalties de la retina i fer-ne el diagnòstic.
Tot i que la teràpia amb cèl•lules mare encara és en plena investigació, ja s’està aplicant en fase de proves i amb molt bons resultats a pacients amb distròfies retinals, retinosi pigmentària i DMAE. Estudis en fase I en humans semblen demostrar que l’ús de cèl•lules mare embrionàries o pluripotencials aconsegueix millorar l’agudesa visual dels pacients, ja que permet reemplaçar les cèl•lules fotoreceptores i/o de l’epiteli pigmentari que no es regeneren i que són fonamentals per a la visió. Aquest resultat s’aconsegueix modificant genèticament les cèl•lules mare, extretes de la pell o d’altres parts de l’ull, perquè puguin desenvolupar la mateixa funció que les cèl•lules danyades.
La DMAE (degeneració macular associada a l’edat) és un dels grans reptes actuals de l’oftalmologia. Sabem que n’hi ha dos tipus: la forma seca i la forma humida. La seca és la que pateixen els pacients que, lentament, perden visió. S’ha demostrat que amb tractaments amb antioxidants i vitamines es pot reduir aquesta pèrdua visual, tot i que el fre a l’evolució no és gaire espectacular. Les formes humides, anomenades així perquè es produeix líquid a la màcula, són les més destructives, i el tractament actual combina la teràpia fotodinàmica, que es va començar a aplicar fa uns quants anys, amb altres tractaments, ja que sembla clar que així s’obtenen efectes més positius. Cada any apareixen noves opcions per intentar avançar en la lluita contra aquesta malaltia.
Potser t'interessa
IMO Institut de Microcirurgia Ocular
Josep María Lladó, 3
08035 Barcelona
Tel: 934 000 700
E-mail: informacion@imo.es
Veure mapa a Google Maps
En cotxe
Coordenades navegador GPS:
41º 24’ 38” N – 02º 07’ 29” E
Sortida 7 de la Ronda de Dalt (costat muntanya). La clínica compta amb un aparcament de més de 200 places.
En bus
Autobús H2: Rotonda de Bellesguard, parada 1540
Autobús 196: Josep Maria Lladó-Bellesguard, parada 3191
Autobusos H2, 123, 196: Ronda de Dalt – Bellesguard, parada 0071
Com arribar a IMO des de:
IMO Grup Miranza Madrid
C/ Valle de Pinares Llanos, 3
28035 Madrid
Tel: 910 783 783
Veure mapa en Google Maps
Transport públic
Metre Lacoma (línia 7)
Autobusos:
- Línies 49 i 64, parada “Senda de l’Infant”
- Línia N21, parada “Metre Lacoma”
Horaris
Atenció al pacient: de dilluns a divendres, de 8 h a 21 h
IMO Grup Miranza Andorra
Av. de les Nacions Unides, 17
AD700 Escaldes-Engordany, Andorra
Tel: (+376) 688 55 44
Veure mapa a Google Maps
IMO Manresa
C/ Carrasco i Formiguera, 33 (Baixos)
08242 – Manresa
Tel: 938 749 160
Veure el mapa a Google Maps
Transport públic
FGC. Línia R5 i R50 direcció Manresa. Estació: Baixador de Manresa
Horaris
De dilluns a divendres de 9:00 a 19:00h